密度泛函理论(Density Functional Theory,简称DFT)是一种计算量子力学方法,用于研究多电子体系的电子结构,特别是分子和凝聚态物理学中的固体。DFT的核心思想是,一个多电子体系的基态性质可以通过其电子密度来唯一确定,而不是通过波函数。这一理论的基础是Hohenberg-Kohn定理,它证明了电子密度与体系的基态能量之间存在一一对应的关系。
DFT的主要特点:1.电子密度为中心:DFT将电子密度作为基本变量,而不是传统的波函数。这使得计算更加直观和易于处理。
2.计算效率:与波函数方法(如Hartree-Fock方法和后Hartree-Fock方法)相比,DFT通常需要较少的计算资源,使其适用于大型体系的计算。
3.适用范围广泛:DFT可以应用于从分子到固体的多种体系,包括金属、半导体和绝缘体。
4.近似的处理:由于DFT中交换-相关能量的确切形式未知,通常需要使用近似的交换-相关泛函。这些泛函的准确性直接影响计算结果的可靠性。
5.可扩展性:DFT方法可以很容易地与其他方法(如多体微扰理论、动态平均场理论等)结合,以提高对特定体系的描述能力。
DFT应用领域:材料科学:用于研究材料的电子结构、晶体结构、光学性质、磁性等。例如,通过计算材料的能带结构和态密度,了解材料的导电性和半导体特性;预测材料的晶体结构和晶格常数,为材料的合成和制备提供理论指导;研究材料的磁性起源和磁相互作用,设计新型磁性材料。化学:在化学反应机理研究、分子结构与性质关系的探讨、催化过程的理解等方面发挥着重要作用。例如,计算化学反应的过渡态和反应路径,揭示反应的微观机制;研究分子的电子云分布和化学键性质,解释分子的化学活性和反应选择性;模拟催化剂表面与反应物分子的相互作用,优化催化剂的设计。
物理学:在凝聚态物理领域,用于研究固体的各种性质,如电子输运、超导性、铁电性等。例如,通过计算电子-声子相互作用,理解材料的超导机制;研究铁电材料的极化性质和相变过程,为开发高性能铁电材料提供理论支持。
Hohenberg-Kohn定理:Hohenberg-kohn定理是密度泛函理论的基石,为用电子密度来描述多电子体系的基态性质提供了理论依据,以下是其详细介绍:
第一定理:对于一个处在外部势场Vext(r)中的多电子体系,其基态电子密度ρ(r)是唯一确定的,并且反过来,外部势场Vext(r)(除了一个常数项)也由基态电子密度ρ(r)唯一确定。这意味着体系的所有性质都可以由电子密度ρ(r)来确定,因为外部势场决定了体系的哈密顿量,进而决定了体系的所有量子力学性质。
第二定理:体系的基态能量Egs是电子密度ρ(r)的泛函E[ρ],并且在满足粒子数守恒∫ρ(r)dr=N(N为体系电子总数)的条件下,当电子密度取到正确的基态密度ρgs(r)时,能量泛函E[ρ]达到最小值,这个最小值就是体系的基态能量Egs,即Egs=E[ρgs]=minE[ρ]。
Hohenberg-Kohn定理从理论上证明了可以通过寻找合适的电子密度泛函来计算多电子体系的基态能量和其他性质,而无需像传统的量子力学方法那样直接处理多电子波函数。由于电子密度是一个三维空间的函数,相比多电子波函数(其变量个数随电子数增加而急剧增加),处理起来要简单得多,这为发展高效的计算方法提供了可能,使得密度泛函理论在处理大量原子的复杂体系时具有很大的优势,在物理、化学、材料科学等领域得到了广泛的应用。
第一定理的证明:假设存在两个不同的外部势场Vext1(r)和Vext2(r),它们产生了相同的基态电子密度ρ(r)。通过构造一个新的哈密顿量,并利用变分原理,可以证明这两个外部势场只能相差一个常数,从而证明了基态电子密度对外部势场的唯一确定关系(除常数项外)。
第二定理的证明:利用变分原理,将体系的能量表示为电子密度的泛函,并证明在满足粒子数守恒的条件下,当电子密度取基态密度时,能量泛函达到最小值。具体证明过程中需要用到一些数学技巧和对量子力学基本原理的深入理解。
需要注意的是,Hohenberg - Kohn 定理只是从原理上说明了电子密度泛函的存在性和重要性质,但并没有给出具体的能量泛函形式。在实际应用中,需要对能量泛函进行近似,如常见的局域密度近似(LDA)、广义梯度近似(GGA)等,以便进行具体的计算。
Khon-Sham方程:Kohn-Sham方程是密度泛函理论中的核心方程,它为计算多电子体系的基态性质提供了一种有效的方法。基于Hohenberg-Kohn定理,将多电子体系的能量表示为电子密度的泛函。然后,引入一个无相互作用的参考体系,其电子密度与真实多电子体系的电子密度相同。通过变分原理,对能量泛函求极值,得到Kohn-Sham方程,具体形式为:

其中:
ψi(r)是第i个Kohn-Sham轨道波函数
ϵi是对应的轨道能量
Veff(r)是有效势能,它包括几个部分:
1.外部势能Vext(r),例如原子核产生的势能。
2.电子间的库仑势能VH(r),即Hartree势能,描述电子间的直接库仑相互作用。
3.交换关联势能Vxc(r),描述电子间的交换和关联作用。
有效势能Veff(r)可以具体写为:

VH(r)是Hartree势能,由电子密度n(r)决定:

Vxc(r)是交换关联势能,它是电子密度的泛函,通常需要通过近似来计算。Kohn-Sham 方程将多电子问题转化为一组单粒子方程,使得我们可以通过求解这些单粒子方程来获得多电子体系的基态能量和电子密度等性质。它在密度泛函理论的实际应用中起着关键作用,为计算材料的电子结构、化学键、光学性质等提供了重要的理论基础。通过选择合适的交换-关联势近似,Kohn-Sham方程能够在一定程度上准确描述各种多电子体系的性质,在物理、化学、材料科学等领域得到了广泛的应用。
尽管DFT在处理大型体系时具有优势,但它也有局限性,特别是在处理强相关电子体系和范德华作用方面。近年来,研究者们致力于开发更精确的交换关联泛函,以更好地描述电子间的复杂相互作用。由于交换关联势的精确形式未知,因此通常需要使用近似的交换关联泛函。这些泛函的准确性直接影响计算结果的可靠性。例如,Becke88-Perdew86 (BP86) 、Perdew–Wang91 (PW91)、Becke88–Perdew–Wang91(BPW91) 和 BLYP (Becke for exchange, with Lee–Yang–Parr for correlation)等是目前最常用的几种泛函。这些泛函通过引入梯度校正项来改进对分子结合能的描述,从而提高了计算的准确性。
DFT计算流程:自洽场迭代:在实际计算中,通常采用自洽场(SCF)迭代方法来求解密度泛函理论的方程。首先,给定一个初始的电子密度分布,然后计算出相应的哈密顿量,进而求解薛定谔方程得到电子的波函数和能量。根据得到的波函数,更新电子密度分布,再重新计算哈密顿量,如此循环迭代,直到电子密度和能量收敛到给定的精度要求。
数值计算方法:为了实现密度泛函理论的计算,需要采用数值方法来离散化和求解相关的方程。常见的方法有平面波基组方法、原子轨道基组方法、赝势方法等。平面波基组方法具有计算效率高、易于并行化等优点,适用于周期性体系;原子轨道基组方法则能更准确地描述原子附近的电子行为,适用于分子体系;赝势方法通过引入赝势来替代原子核与内层电子对价电子的作用,大大减少了计算量。DFT计算内容:1.本征值与波函数基态电子密度:通过求解Kohn-Sham方程,得到体系的基态电子密度分布ρ(r),它描述了电子在空间中的概率分布情况,直观地展现了电子在原子、分子或固体中的分布特征,例如可以显示电子在化学键区域的聚集以及在原子周围的分布规律。
单粒子能级和波函数:Kohn-Sham方程的解给出了单粒子能级ϵi和相应的波函数ψi(r)。单粒子能级可以理解为电子所处的能量状态,而波函数则提供了电子的量子态信息,通过波函数可以计算出电子的各种物理量的期望值。
2.系统能量基态能量:根据Hohenberg-Kohn 第二定理,通过对能量泛函E[ρ]进行计算,并在满足粒子数守恒的条件下进行变分,得到体系的基态能量。基态能量是体系在最低能量状态下的能量值,它是一个非常重要的物理量,例如可以通过基态能量来比较不同结构的稳定性,基态能量越低,体系越稳定。
总能量:DFT计算得到的总能量包括电子的动能、电子-原子核之间的相互作用能、电子 - 电子之间的相互作用能(包括Hartree能和交换-关联能)等。通过对总能量的分析,可以了解体系中各种相互作用对体系能量的贡献。
能量差:在研究化学反应、材料相变等过程中,计算不同状态之间的能量差是非常重要的。例如,通过计算反应物和产物的能量差,可以得到反应的焓变,从而判断反应是否能够自发进行;通过计算材料在不同晶相下的能量差,可以确定材料的稳定晶相。
3.电荷与磁性电荷分布和键级:由电子密度分布可以进一步分析体系的电荷分布情况,例如可以计算原子上的电荷分布,了解电子在不同原子之间的转移情况,从而判断化学键的类型和强度。键级是衡量化学键强度的一个重要指标,通过DFT计算可以得到键级的数值,键级越大,化学键越强。
磁性:对于具有磁性的材料,DFT可以计算其磁矩、磁性结构等。通过考虑电子的自旋自由度,计算不同自旋状态下的能量和电子结构,从而研究材料的磁性起源和磁性性质,例如可以确定材料是铁磁性、反铁磁性还是顺磁性等。
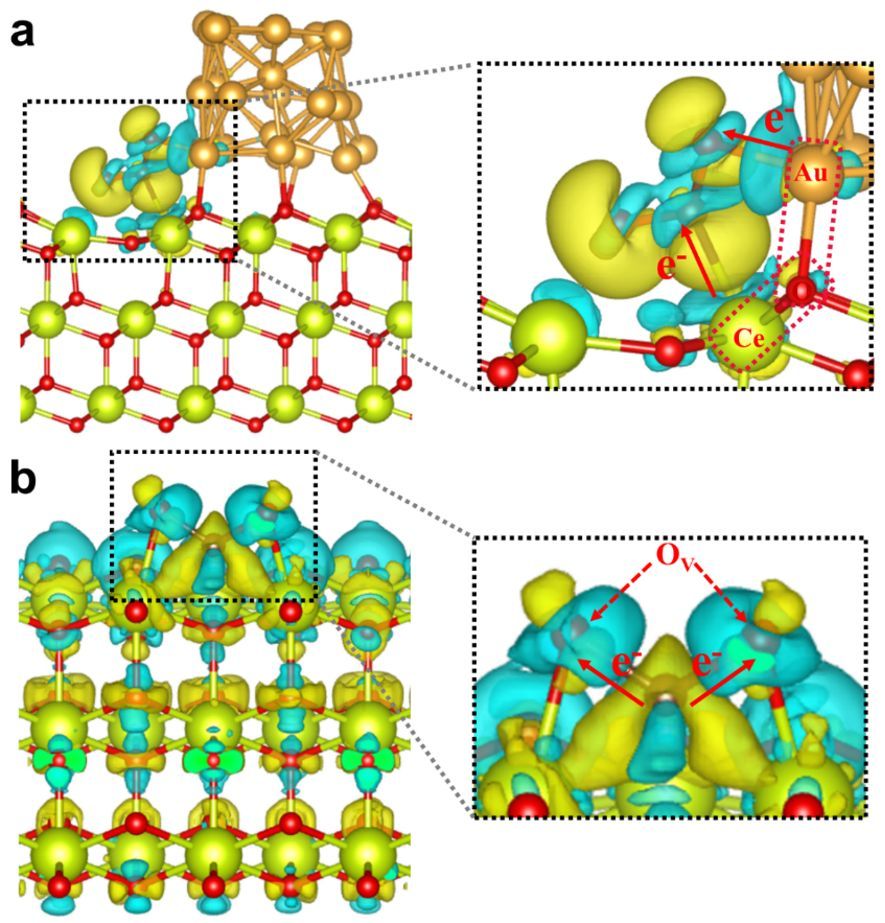
差分电荷

静电势
4.结构性质原子坐标优化:DFT计算可以用于优化分子或晶体的结构,即通过调整原子的坐标,使得体系的总能量达到最小。在优化过程中,计算机会根据能量对原子坐标的导数(即原子所受的力)来逐步调整原子的位置,直到原子所受的力小于某个阈值,此时得到的结构即为体系的稳定结构。
晶格参数优化:对于晶体材料,除了优化原子坐标外,还可以优化晶格参数,如晶格常数、晶胞角度等。通过改变晶格参数并计算相应的总能量,找到使总能量最低的晶格参数,从而得到晶体的稳定晶格结构。结构优化对于准确研究材料的性质非常重要,因为不同的结构可能导致材料性质的巨大差异。

晶体结构
5.催化性质吸附性质研究:通过计算吸附能来判断反应物或中间体在催化剂表面的吸附强弱和方式。例如,在研究二氧化碳加氢反应时,计算二氧化碳和氢气在催化剂表面的吸附能,能了解反应起始的难易程度,以及不同催化剂对反应物的吸附选择性。
反应机理探究:确定催化反应的可能路径和中间体,计算每一步反应的活化能和反应热,从而揭示反应的微观机理。以甲醇合成反应为例,借助DFT 计算可以详细分析一氧化碳和氢气在催化剂表面转化为甲醇的各个步骤,明确速率控制步骤,为催化剂的设计提供理论依据。
催化剂活性预测:结合吸附性质和反应机理的研究结果,对催化剂的活性进行预测。例如,对于一系列不同金属掺杂的催化剂,通过DFT 计算评估它们对特定反应的催化活性,筛选出具有潜在高活性的催化剂,减少实验筛选的工作量和成本。
催化剂稳定性分析:考察催化剂在反应条件下的结构稳定性和抗中毒能力。比如,研究催化剂表面在高温、高压或存在杂质的情况下,是否会发生结构变化或被杂质吸附而失活,为催化剂的实际应用提供稳定性方面的信息。

反应路径与吉布斯自由能

态密度

反应过渡态
6.电池性质电极材料结构与稳定性:通过DFT 计算可以优化电极材料的晶体结构,预测其在不同条件下的稳定性。例如,对于锂离子电池的正极材料 LiCoO₂,计算可以确定其在充放电过程中晶体结构的变化情况,以及可能出现的相转变,为材料的实际应用提供理论依据。
电子结构与导电性:分析电极材料的电子结构,如能带结构、态密度等,能够了解材料的导电性能。以石墨烯为例,DFT 计算表明其具有独特的二维电子结构,载流子迁移率高,这使其成为一种有潜力的电池电极材料或添加剂,可用于提高电极的导电性和电池的充放电性能。
离子扩散行为:计算离子在电极材料中的扩散路径和扩散能垒,有助于理解电池的充放电动力学。例如,在研究锂离子电池负极材料硅时,DFT 计算可以揭示锂离子在硅晶格中的扩散机制,发现锂离子在某些晶向的扩散能垒较低,从而为优化材料结构以提高锂离子扩散速率提供方向。
离子电导率:通过计算电解质中离子的迁移率和扩散系数,评估电解质的离子电导率。例如,对于固态电解质Li₃PS₄,DFT 计算可以确定锂离子在其晶体结构中的迁移路径和活化能,进而预测其离子电导率。研究发现,通过适当的掺杂可以改变电解质的晶体结构,降低锂离子的迁移能垒,提高离子电导率。
与电极的相容性:研究电解质与电极材料之间的界面相互作用,评估两者的相容性。在锂离子电池中,电解质与电极之间的界面稳定性对电池的性能和循环寿命至关重要。DFT 计算可以分析电解质与电极表面的吸附作用、电荷转移情况以及界面处的化学反应,为选择合适的电解质和电极材料组合提供理论指导,以减少界面副反应,提高电池的稳定性。
电极反应过程:模拟电池充放电过程中电极材料与电解质之间的化学反应,确定反应的中间体和反应路径。以锂- 空气电池为例,DFT 计算可以研究氧气在电极表面的吸附和还原过程,以及锂离子与氧物种之间的相互作用,揭示锂 - 空气电池放电过程中形成的锂过氧化物等中间产物的生成机制,为改善电池的充放电性能和循环稳定性提供理论支持。
电池热效应:计算电池反应的焓变和熵变,评估电池的热效应。在锂离子电池中,充放电过程中的热效应会影响电池的性能和安全性。通过DFT 计算可以预测不同电极材料和电解质组合下电池反应的热效应,为电池的热管理设计提供理论依据,有助于提高电池的安全性和使用寿命。

离子迁移

离子嵌入
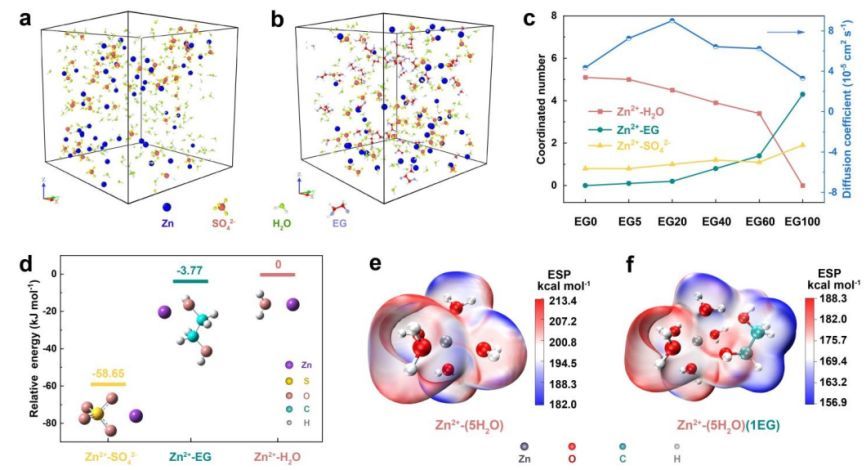
电解液
7.半导体性质能带结构:通过求解Kohn - Sham 方程,DFT 可以给出半导体的能带结构,包括导带、价带以及它们之间的带隙。例如,对于常见的半导体硅,DFT 计算能够准确得出其间接带隙的大小,以及导带底和价带顶的位置和形状,从而了解电子在晶体中的能量分布和跃迁情况。
态密度:DFT 计算可以得到半导体的电子态密度,它表示在不同能量状态下电子出现的概率。通过分析态密度,能了解半导体中电子的分布情况以及不同原子轨道对电子态的贡献。比如在氮化镓(GaN)半导体中,通过态密度分析可以明确镓(Ga)和氮(N)原子的轨道电子在价带和导带中的分布情况,进而理解其光学和电学性质。
吸收光谱:基于DFT 计算得到的电子结构,可以进一步计算半导体的光学吸收光谱。当光子能量被电子吸收后,电子会从价带跃迁到导带,通过计算这种跃迁的概率和能量变化,能得到吸收光谱。以氧化锌(ZnO)为例,DFT 计算可以预测其在紫外 - 可见光区域的吸收特性,为其在光电器件中的应用提供理论依据。
激子效应:激子是由受光激发而形成的束缚态的电子- 空穴对,在一些半导体中激子效应较为显著。DFT 可以通过计算电子 - 空穴之间的库仑相互作用来研究激子的形成、结合能以及激子在半导体中的传输等性质。例如在二维半导体过渡金属二硫化物(TMDCs)中,DFT 计算有助于深入理解激子的特性,为其在光电器件中的应用提供支持。
载流子迁移率:DFT 可以计算半导体中载流子(电子和空穴)的有效质量,进而通过理论模型估算载流子迁移率。有效质量反映了载流子在晶体中受到晶格势场作用的运动难易程度。例如,对于碳化硅(SiC)半导体,通过 DFT 计算其电子和空穴的有效质量,能够预测其在不同晶向的载流子迁移率,为设计高性能的 SiC 电子器件提供参考。
扩散系数:借助DFT 计算可以研究载流子在半导体中的扩散行为,得到扩散系数。这对于理解半导体器件中的电流传输、杂质扩散等过程非常重要。比如在研究有机半导体中的载流子传输时,DFT 计算可以帮助分析载流子在分子间的扩散机制,为提高有机半导体器件的性能提供指导。
表面态:半导体表面由于原子排列的不完整性会形成表面态,影响半导体的性能。DFT 计算可以研究半导体表面的电子结构和表面态的分布情况。例如,对于硅半导体表面,通过 DFT 计算可以分析不同表面重构方式下的表面态,了解其对表面吸附、催化等性质的影响。
界面特性:在半导体异质结等结构中,界面特性至关重要。DFT 计算可以研究不同半导体材料界面处的电子结构、电荷转移以及界面态等性质。以砷化镓(GaAs)/ 氮化镓(GaN)异质结为例,DFT 计算能够揭示界面处的能带排列、电荷分布情况,为设计高性能的异质结光电器件提供理论基础。

能带结构与轨道分布

原子迁移

缺陷能级

功函数

光学性质
https://www.huasuankeji.com/news/?p=328401